原子吸收光谱(综述)
贡献者: 待更新
(本文根据 CC-BY-SA 协议转载自原搜狗科学百科对英文维基百科的翻译)

原子吸收光谱(AAS)和原子发射光谱(AES)是一种利用自由原子对气态光辐射(光)的吸收,定量测定化学元素的光谱分析方法。原子吸收光谱是以自由金属离子对光的吸收为基础的。
在分析化学中,该技术用于确定待分析样品中特定元素(被分析物)的浓度。原子吸收光谱法可用于测定溶液中 70 多种不同的元素,也可以通过电热蒸发直接测定固体样品中的元素,用于药理学、生物物理学,考古学和毒理学研究。
原子发射光谱学最初被用作分析技术,其基本原理是由德国海德堡大学的教授 Robert Wilhelm Bunsen 和 Gustav Robert Kirchhoff 在 19 世纪下半叶确立的。[1]
原子吸收光谱的现代形式主要是在 20 世纪 50 年代由一组澳大利亚化学家发展起来的。他们由澳大利亚墨尔本联邦科学与工业研究组织(CSIRO)化学物理部门的的 Alan Walsh 爵士领导。[2][3]
原子吸收光谱法在化学的不同领域有许多用途,如临床分析生物液体和组织中的金属,如全血、血浆、尿液、唾液、脑组织、肝脏、头发、肌肉组织、精液,在一些制药过程中,在最终药物产品中残留的微量催化剂,以及分析水中的金属含量。
1. 原则
原子吸收光谱技术利用样品的原子吸收光谱来评估样品中特定分析物的浓度。它需要具有已知分析物含量的标准来建立测量吸光度和分析物浓度之间的关系,因此原子吸收光谱依赖于 Beer-Lambert 定律。
2. 原子吸收光谱仪器
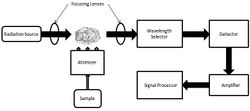
为了分析样品的原子成分,必须对样品进行雾化。现在最常用的雾化器是火焰和电热(石墨管)雾化器。用光辐射照射原子,辐射源可以是元素特异性线辐射源,也可以是连续辐射源。然后,辐射通过单色仪,单色仪将特定元素的辐射与辐射源发射的所有其他辐射分离开来,最终由检测器进行测量。
2.1 雾化器
原子吸收光谱法中最古老和最常用的雾化器是火焰,主要是温度约为 2300℃的空气-乙炔火焰以及温度约为 2700℃的一氧化二氮[3] -乙炔火焰。另外,后一种火焰提供的雾化环境还原性更强,非常适合对氧具有高亲和力的分析物。

液体或溶液样品通常与火焰雾化器一起使用。样品溶液由气动分析喷雾器吸入,转化为气溶胶,并被引入喷雾室,只有最细的气溶胶液滴(< 10 微米)进入火焰,在喷雾室中与火焰气体混合。这种调节过程导致只有约 5%的吸入样品溶液到达火焰,但它也保证了相对较高的干扰自由度。
喷雾室的顶部是一个燃烧器头,它产生横向较长的火焰(通常为 5-10 cm)并且宽度只有几毫米。辐射光束穿过火焰的最长轴,可以通过调节火焰气体的流速来产生最高浓度的自由原子。还可以调整燃烧器高度,使辐射光束通过火焰中原子云密度最高的区域,产生最高的灵敏度。
在火焰中发生的过程包括去溶剂化(干燥)阶段,在该阶段溶剂蒸发,干燥的样品纳米颗粒保留,蒸发(转移到气相)阶段,在该阶段固体颗粒转化成气体分子,雾化阶段,气体分子解离成自由原子,然后电离(取决于分析物原子的电离电势和特定火焰中可用的能量),其中原子可以部分转化为气态离子。
如果校准标准和样品中的分析物的相转移程度不同,则这些阶段中的每一个都包括干扰风险。这种情况下我们通常不希望发生电离,因为它减少了可用于测量的原子数量,即灵敏度。
在火焰原子吸收光谱法中,样品被吸出时会产生稳态信号。该技术通常用于 $mg L^{-1}$ 范围内的测定,对于某些元素来说可扩展至 $\mu g L^{-1}$。
电热雾化器
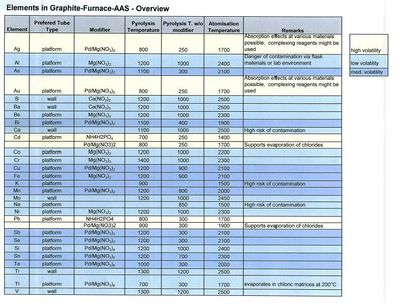
使用石墨管雾化器的电热原子吸收法是俄罗斯圣彼得堡理工学院的 Boris V. L’vov 于 20 世纪 50 年代末首创的[4],德国多特蒙德光谱化学和应用光谱学研究所(ISAS)的汉斯·马斯曼也同时进行来研究。[5]

尽管多年来使用了各种各样的石墨管设计,但现在石墨管的尺寸通常为长度 20–25 mm、内径 5-6 mm。利用这种技术,可以直接分析液体/溶液、固体和气体样品。将测量一定体积(通常为 10–50 $\mu L$)或称重一定质量(通常约为 1 mg)的固体样品引入石墨管并进行温度程序。电热雾化过程通常包括几个阶段,如干燥——溶剂蒸发;热解——大部分基质成分被去除;雾化——分析物元素释放到气相中;清洗——在高温下清除石墨管中的最终残留物。
采用低压大电流电源,通过石墨管的欧姆电阻加热石墨管;各个阶段的温度可以被非常紧密地控制,而且各个阶段之间的温度梯度有利于样品组分的分离。管子可以横向加热,也可以纵向加热,前者的优点是其长度上的温度分布更加均匀。Walter Slavin 在 Boris L 'vov 研究的基础上提出的所谓稳定温度平台炉(STPF)概念,使原子吸收光谱法基本不受干扰。这一概念的主要组成部分是样品的雾化在插入石墨管(L'vov 平台)的石墨平台发生而不是在管壁发生,这样可以延迟雾化,直到雾化器中的气相达到稳定温度;使用化学改性剂,以将分析物稳定到足以除去大部分基质组分的热解温度;并对吸光度随时间变化的瞬态吸收信号进行积分,而不是用峰高吸光度进行量化。
在原子吸收光谱法中,产生的瞬态信号面积与引入石墨管的分析物质量(不是其浓度)成正比。这种技术的优点是可以直接分析固态、液态或气态任何种类的样品。其灵敏度比火焰原子吸收光谱法高 2-3 个数量级,因此可以在低 $\mu g L^{-1}$ 情况下进行测量(在典型样品体积为 20 $\mu g $ 的情况下),也可以在 $\mu g L^{-1}$ 范围测量(在典型样品质量为 2 mg 的情况下)。原子吸收光谱法具有很高的抗干扰性,因此原子吸收光谱法可被认为是目前测定复杂基体中痕量元素的最强有力的技术
专业雾化技术
虽然火焰雾化器和电热雾化器是最常见的雾化技术,但也有几种其他雾化方法被用于特殊用途。[6][7]
辉光放电雾化器
辉光放电装置是一种多功能光源,因为它可以同时引入和雾化样品。辉光放电发生在 1-10 torr 的低压氩气气氛中。在这种气氛中,一对电极施加 250 至 1000 伏的 DC 电压,将氩气分解成带正电荷的离子和电子。在电场的影响下,这些离子被加速进入包含样品的阴极表面,轰击样品,并通过所谓的溅射过程导致中性样品原子喷射。这种放电产生的原子蒸汽由离子、基态原子和部分受激原子组成。当受激原子回到基态时,会发出低强度的辉光,这项技术因此得名。
辉光放电雾化器对样品的要求是样品必须是电导体。因此,辉光放电雾化器最常用于金属和其他导电样品的分析。然而,通过适当的改进,可以通过让样品与导体(如石墨)混合来分析液体样品和不导电材料。
氢化物雾化器
氢化物发生技术是专门研究特定元素溶液的技术。该技术提供了一种将含有砷、锑、硒、铋和铅的样品引入气相雾化器的方法。与其他方法相比,氢化物雾化器将检测限提高了 10 到 100 倍。氢化物发生是通过将样品的酸化水溶液加入 1% 硼氢化钠水溶液中进行的,所有水溶液都装在玻璃容器中。反应产生的挥发性氢化物被惰性气体吹入雾化室,在那里发生分解。这一过程形成了被分析物的雾化形式,然后可以通过吸收或发射光谱测定法进行测量。
冷蒸汽雾化器
冷蒸汽技术是一种雾化方法,只限于测定汞,因为汞是环境温度下唯一具有足够大蒸汽压的金属元素。因此,它在测定样品中的有机汞化合物及其在环境中的分布方面具有重要用途。该方法首先将 Hg 用硝酸和硫酸的氧化为 $Hg^2+$,然后用氯化锡(II)还原 $Hg^2+$。然后,通过使惰性气体流鼓泡通过反应混合物,将汞吹入长通吸收管。通过测量该气体在 253.7 nm 处的吸光度来确定汞的浓度。这种技术的检测限在十亿分之一的范围内,是一种优秀的汞检测雾化方法。
2.2 辐射源
我们必须区分线源原子吸收法和连续源原子吸收法。在经典的最小二乘原子吸收光谱法中,正如 Alan Walsh 提出的那样,[8] 原子吸收光谱测量所需的高光谱分辨率由辐射源本身提供,辐射源以比吸收线窄的线的形式发射分析物的光谱。连续光源,如氘灯,仅用于背景校正。这种技术的优点是测量原子吸收光谱只需要一个中分辨率单色仪;然而,它的缺点是,通常每个必须确定的元素都需要单独的灯。相比之下,在铯原子吸收光谱法中,所有元素都使用单个灯,在感兴趣的整个光谱范围内发射连续光谱。显然,这项技术需要高分辨率单色仪,这将在后面讨论。
空心阴极灯

空心阴极灯(HCL)是原子吸收光谱法中最常见的辐射源。在密封的灯里面,充有低压氩气或氖气,有一个圆柱形金属阴极(由被测元素的金属或合金化合物构成)和阳极。在阳极和阴极之间施加高电压,导致填充气体电离。气体离子向阴极加速,并且在撞击阴极时,溅射阴极材料,阴极材料在辉光放电中被激发,发射出被溅射材料的辐射,即被测元素的辐射。在大多数情况下,使用单元素灯,其中阴极主要由目标元素的化合物压制而成。多元素灯可在阴极中用目标元素化合物的组合压制而成。多元素灯的灵敏度略低于单元素灯,必须仔细选择元素组合,以避免光谱干扰。大多数多元件灯结合的元素较少,如 2-8 个。原子吸收光谱仪一般只有 1-2 个空心阴极灯,在自动多元件光谱仪中,通常可以提供 8-12 个灯。
无电极放电灯
无电极放电灯(EDL)在石英灯泡中含有少量金属或盐形式的分析物以及低压惰性气体,通常为氩气。灯泡被插入产生电磁射频场的线圈中,导致灯中的低压电感耦合放电。EDL 的辐射比 HCL 的辐射高,光谱线宽一般较窄,但 EDL 需要单独的电源,可能需要较长的时间来稳定。
氘灯
在线光源原子吸收光谱法使用氘空心阴极灯(HCL)、氢空心阴极灯(HCL)和氘放电灯进行背景校正。[9] 这些灯发射的辐射强度随着波长的增加而显著降低,因此它们只能在 190 nm 到 320 nm 之间的波长范围内使用。
氙灯连续辐射源

当氙灯连续辐射源用于原子吸收光谱法时,必须使用高分辨率分光器,这将在后面讨论。此外,灯在 190 nm 至 900 nm 的整个波长范围内发射的辐射强度必须至少比典型空心阴极灯(HCL)高一个数量级。为了满足这些要求,研制了一种特殊的高压氙气短弧灯。
2.3 分光器
如上所述,用于线光源原子吸收光谱法的中分辨率分光器和用于连续光源吸收光谱法的高分辨率分光器有所不同。分光器包括光谱分类装置(单色仪)和探测器。
线光源原子吸收光谱分光器
在线光源原子吸收光谱分光系统中,测量原子吸收所需的高分辨率由辐射源的窄线发射提供,单色仪只需从灯发射的其他辐射中分辨出分析线。这通常可以通过 0.2-2 nm 的带通来实现,即中等分辨率单色仪。使线光源原子吸收光谱法元件特定的另一个特征是初级辐射的调制和调谐到相同调制频率的选择性放大器的使用,如 Alan Walsh 所假设的那样。这样,可以排除任何(未调制的)辐射,例如雾化器发出的辐射,这对线光源原子吸收光谱法是必不可少的。Littrow 的简单单色计以及(更好的)Czerny-Turner 改良的简单单色仪通常用于线光源原子吸收光谱法。光电倍增管是线光源原子吸收光谱法中最常用的检测器,尽管固态探测器可能是首选,因为它们具有更好的信噪比。
连续光源原子吸收光谱分光器
当连续辐射源用于原子吸收光谱测量时,使用高分辨率分光器是必不可少的。分辨率必须等于或优于原子吸收线的半宽度(约 2 pm),以避免校准图的灵敏度和线性损失。高分辨率连续光源原子吸收光谱分光器的研究是由是由美国的 O'Haver 和 Harnly 团队率先开展的,他们也为这项技术开发了(迄今为止)唯一的同步多元素原子吸收光谱仪。然而,当 Becker Ross 团队在德国柏林建造了一台完全为高分辨率连续光源原子吸收光谱设计的光谱仪时,这一突破才得以实现。21 世纪初,Analytick Jena 公司在 Becker Ross 和 Florek 提出的设计基础上,引进了第一台用于高分辨率连续光源原子吸收光谱的商用设备。这些光谱仪使用紧凑型双单色仪,配有棱镜前单色仪和高分辨率的 Echelle 光栅单色仪。探测器采用 200 像素的线阵电荷耦合器件(CCD)阵列作为探测器。第二个单色仪没有出口狭缝;因此分析线两侧的光谱环境在高分辨率下变得可见。由于通常只有 3–5 个像素用于测量原子吸收,其他像素可用于校正。校正之一是对灯闪烁噪声的校正,它与波长无关,导致测量具有非常低的噪声水平;其他校正用于背景吸收的校正,这将在后面进行讨论。
3. 背景吸收和背景校正
相对较少数量的原子吸收线(与原子发射线相比)和它们的窄宽度(几μm)使得光谱重叠很少;只有已知少数几个例子表明一种元素的吸收线会与另一种元素重叠。相比之下,分子吸收要宽得多,因此一些分子吸收带更有可能会与原子线重叠。这种吸收可能是由样品中含有元素的未解离分子或火焰气体引起的。我们必须将显示出明显精细结构的二原子分子光谱和不显示这种精细结构的较大(通常是三原子)分子的光谱区分开来。背景吸收的另一个来源,特别是在原子吸收光谱法中,是当基质在热解阶段不能被充分去除时,初级辐射在雾化阶段产生的颗粒上的散射。
所有这些现象,分子吸收和辐射散射,都会导致人为的高吸收和对样品中分析物浓度或质量的不适当的高(错误的)计算。有几种技术可以用来校正背景吸收,它们在 LS AAS 和 HR-CS AAS 中存在显著差异。
3.1 线光源原子吸收光谱法中的背景校正技术
在原子吸收光谱法中,背景吸收只能用仪器技术校正,而且所有校正都基于两次连续的测量,首先测量总吸收(原子+背景),然后仅测量背景吸收,两次测量的差值就是净原子的吸收。由于这一原因,且由于在光谱仪中使用了额外的设备,背景校正信号的信噪比总是比未校正信号的信噪比低得多。还应该指出,在线光源原子吸收光谱分析中,没有办法校正两条原子线的直接重叠(这种情况很少见)。在线光源原子吸收光谱法中,基本上有三种背景校正技术:
氘背景校正
这是最古老也是最常用的技术,尤其常用于火焰原子吸收光谱法。在这种情况下,使用具有宽发射的独立光源(氘灯)来测量光谱仪出射狭缝整个宽度上的背景吸收。使用单独的灯使这种技术是最不精确的,因为它不能纠正任何结构化的背景。由于氘灯的发射强度在波长 320 nm 以上变得非常微弱,所以它也不能在 320 nm 以上的波长下使用。与弧光灯相比,使用氘空心阴极灯(HCL)是较优选择,因为相比于前者,氘空心阴极灯灯能够更好地与图像匹配。
Smith-Hieftje 背景校正
这项技术(以其发明者的名字命名)基于当施加大电流时空心阴极灯(HCL)发射线的展宽和自反转。用正常的灯电流,即窄发射线测量总吸收,再施加高电流脉冲使其产生自逆转,测量背景总吸收,其在原始波长下的发射量很弱,但在分析线的两侧都有强吸收。这种技术的优点是只使用一个辐射源;缺点之一是大电流脉冲缩短了灯的寿命,而且该技术只能用于相对易挥发的元素,因为只有那些元素具有足够的自逆转能力,以避免发生剧烈的损失。另一个问题是,背景的测量波长与总吸收波长不同,这使得该技术不适合于校正结构化背景。
塞曼效应背景校正
在雾化器(石墨炉)内施加交变磁场,将吸收线分成三个分量,π分量保持在与原始吸收线相同的位置,两个σ分量分别移动到更高和更低的波长。总吸收是在没有磁场的情况下测量的,背景吸收是在磁场开启的情况下测量的。在这种情况下,必须去除π分量,例如使用偏振器,并且σ分量不会与灯的发射轮廓重叠,因此只测量背景吸收。这种技术的优点是总吸收和背景吸收是用相同灯的相同发射曲线测量的,因此任何种类的背景,包括具有精细结构的背景,都可以被精确地校正,除非负责背景的分子也受到磁场的影响。用斩波器作为极化器,可以降低信噪比。塞曼效应背景校正的缺点是分光计的复杂性增加,运行分离吸收线所需的强力磁铁所需的电源也增加了。
3.2 高分辨率连续光源原子吸收光谱中的背景校正技术
在高分辨率连续光源原子吸收光谱中,背景校正是在软件中用数学方法进行的,使用的信息来自探测器像素,而不是来自于原子吸收的测量;因此,与线光源原子吸收光谱法相比,背景校正不需要额外的元件。
使用校正像素的背景校正
已经提到在高分辨率连续光源原子吸收光谱法中使用校正像素消除闪烁噪声。事实上,校正算法消除了在选择用于校正的所有像素处观察到的相同程度的辐射强度的任何增加或减少。这显然还包括由于辐射散射或分子吸收而导致的测量强度的降低,这可以用同样的方法进行校正。由于总吸收和背景吸收的测量以及对后者的校正是严格同步的(与线光源原子吸收光谱法不同),即使是在等原子吸收光谱中观察到的背景吸收的最快变化也不会引起任何问题。此外,由于采用了同样的算法对背景进行校正和消除灯光噪声,与未校正信号相比,背景校正信号的信噪比要高得多,这也与线光源原子吸收光谱法形成了对比。
使用最小二乘法的背景校正
上述技术显然不能对具有精细结构的背景进行校正,因为在这种情况下,每个校正像素的吸光度将不同。在这种情况下,高分辨率连续光源原子吸收光谱提供了一种可能性,可以测量负责背景的分子的校正光谱,并将其存储在计算机中。然后将这些光谱乘以一个系数,以匹配样本光谱的强度,并使用最小二乘法从样本光谱中逐像素和逐光谱地减去。这听起来可能很复杂,但首先,原子吸收光谱法中使用的雾化器的温度下可以存在的二原子分子数量相对较少,其次,计算机在几秒钟内就可以完成校正。同样的算法也可以用来校正两条原子吸收线的直线重叠,使得高分辨率连续光源原子吸收光谱成为唯一可以校正这种光谱干扰的原子吸收光谱技术。
4. 参考文献
- "Robert Bunsen and Gustav Kirchhoff". Science History Institute. Retrieved 20 March 2018..
- McCarthy, G.J. "Walsh, Alan - Biographical entry". Encyclopedia of Australian Science. Retrieved 22 May 2012..
- Koirtyohann, S. R. (1991). "A HISTORY OF ATOMIC ABSORPTION SPECTROMETRY". Analytical Chemistry. 63 (21): 1024A–1031A. doi:10.1021/ac00021a716. ISSN 0003-2700..
- L'vov, Boris (1990). Recent advances in absolute analysis by graphite furnace atomic absorption spectrometry. Spectrochimica Acta Part B: Atomic Spectroscopy. 45. pp. 633–655. Bibcode:1990AcSpe..45..633L. doi:10.1016/0584-8547(90)80046-L..
- "Analytical Methods for Graphite Tube Atomizers" (PDF). agilent.com. Agilent Technologies..
- Harvey, David (2016-05-25). "Atomic Absorption Spectroscopy". chem.libretexts.org..
- "Sample Atomization – Atomic Absorption Spectroscopy Learning Module". blogs.maryville.edu (in 英语). Retrieved 2017-11-02..
- Walsh, Alan; Becker-Ross, Helmut; Florek, Stefan; Heitmann, Uwe (19 January 2006). High-Resolution Continuum Source AAS. Weinheim: Wiley‐VCH Verlag GmbH & Co. KGaA. p. 2. ISBN 9783527307364..
- Rakshit, Amitava. "Basics of Laboratory Safety: Common laboratory rules and regulations". The International Association for Ecology. Intecol. Archived from the original on 27 September 2016. Retrieved 26 September 2016..
友情链接: 超理论坛 | ©小时科技 保留一切权利