聚合酶链反应
编辑聚合酶链式反应(英文:Polymerase chain reaction,缩写:PCR,又称多聚酶链式反应),是一项利用DNA双链复制的原理,在生物体外复制特定DNA片段的核酸合成技术。透过这项技术,可在短时间内大量扩增目的基因,而不必依赖大肠杆菌或酵母菌等生物体。
聚合酶链反应是由凯利·穆利斯(Kary Mullis)于1983年开发,当时他是Cetus公司的雇员,也是1993年诺贝尔化学奖的获得者,它是一种简单,廉价和可靠的方法复制DNA片段,这个概念适用于现代生物学和相关科学的许多领域。 PCR可能是分子生物学中使用最广泛的技术。这种技术被用于生物医学研究,犯罪取证和分子考古学
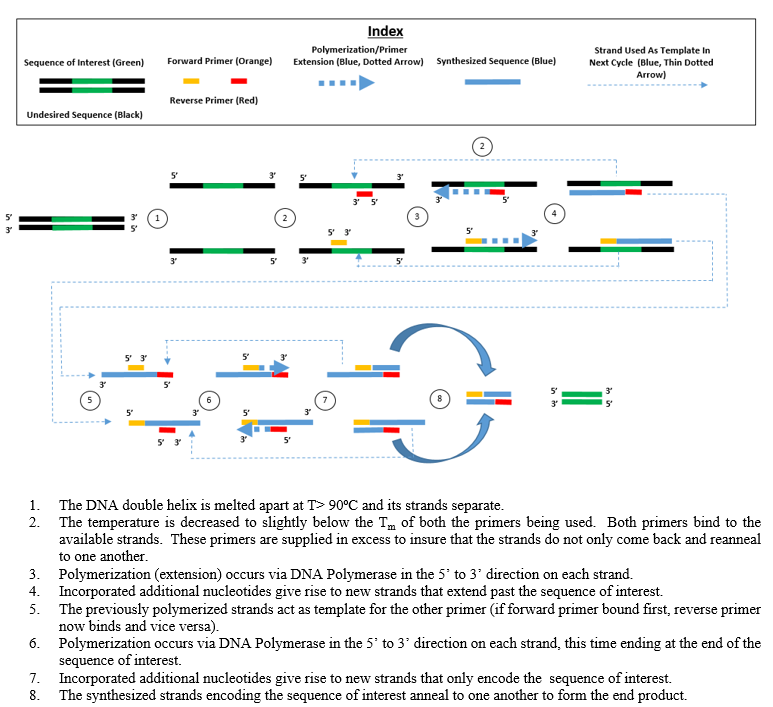
绝大多数聚合酶链反应方法依赖于热循环。热循环将反应物暴露于加热和冷却的重复循环中,以允许不同的温度依赖性反应——具体地说,脱氧核糖核酸融化和酶-驱动DNA复制。聚合酶链反应使用两种主要试剂-引物(引物是短的单链DNA片段,称为寡核苷酸,是目标DNA区域的互补序列)和DNA聚合酶。在PCR反应的第一步,DNA双螺旋结构的两条链在高温下物理分离,这个过程称为脱氧核糖核酸变性。第二步,降低温度,引物与互补的脱氧核糖核酸序列结合。这两条DNA链就变成了模板,以酶促的方式从构成DNA的自由核苷酸中组装出一条新的DNA链。随着聚合酶链反应的进行,产生的DNA本身被用作复制的模板,启动了一个连锁反应,原始的DNA模板是以指数形式放大。
目录编辑
1 原则编辑
PCR扩增DNA链的特定区域(DNA靶)。大多数聚合酶链反应方法扩增长度在0.1至10千碱基对(kbp)之间的DNA片段,尽管一些技术允许扩增高达40 kbp的片段。[1]扩增产物的量由反应中可用的底物决定,随着反应的进行,底物的数量变得有限。[2]
基本的聚合酶链反应设置需要几种组分和试剂,包括包含要扩增的DNA目标区域的DNA模板; DNA聚合酶;一种聚合新DNA链的酶;耐热的 Taq聚合酶特别常见,[3]因为它更有可能在高温DNA变性过程中保持完整;两个DNA引物分别与DNA靶的每条正义链和反义链的 3’末端互补(DNA聚合酶只能与DNA双链区域结合并从双链区域延伸;没有引物,就没有聚合酶可以结合的双链起始位点);[4]预先选择与DNA靶区互补的特异性引物,通常在实验室定制或从商业生化供应商处购买;脱氧核苷三磷酸,或dNTPs(有时称为“脱氧核苷酸三磷酸”;核苷酸含有三磷酸基团),DNA聚合酶从中合成新的DNA链的构建模块;a缓冲溶液为DNA聚合酶的最佳活性和稳定性提供合适的化学环境;二价 阳离子通常为镁或锰离子;其中镁离子是最常见的,但是Mn2+可作为高锰酸盐用于PCR介导的DNA诱变, 锰离子浓度会增加DNA合成过程中的错误率;[5]其中单价阳离子通常为钾离子
PCR反应通常在10-200 μL体积内进行,在小反应管中(0.2-0.5 μL体积)放入热循环仪中。热循环仪加热和冷却反应管,以达到反应中每个步骤所需的温度(见下文)。许多现代热循环仪利用珀耳帖效应,该效应允许通过简单地逆转电流来加热和冷却PCR管。薄壁反应管允许良好的导热性,以实现快速热平衡。大多数热循环仪都有加热的盖子,以防止反应管顶部冷凝。没有加热盖的老式热循环仪需要在反应混合物顶部涂一层油,或者在管内涂一团蜡。
1.1 程序
通常,PCR由一系列20-40次重复的温度变化组成,称为热循环,每个循环通常由两个或三个离散的温度步骤组成(见下图)。循环通常在非常高的温度下进行单个温度步骤(>90 degrees Celsius (194 degrees Fahrenheit)),然后在最后保持一次,以便最终产品扩展或短暂存储。在每个循环中使用的温度和时间取决于各种参数,包括用于DNA合成的酶、反应中二价离子和dNTPs的浓度以及引物的熔解温度(Tm)。[6]大多数聚合酶链反应方法共有的步骤如下:
- 初始化:此步骤仅适用于需要热激活的DNA聚合酶热启动聚合酶链反应。它包括将反应室加热至94–96 °C (201–205 °F),或 98 °C (208 °F),如果使用极端耐热的聚合酶,然后保持1-10分钟。
- 变性:该步骤是第一次常规循环活动,包括将反应室加热至94-98°C(201-208°F) 持续20-30秒。通过破坏互补碱基之间的氢键,导致脱氧核糖核酸熔解或变性,产生两个单链DNA分子。
- 退火:在下一步中,反应温度降低到50-65℃(122-149°F)持续20-40秒,使每个单链DNA模板上的引物退火。反应混合物中通常包含两种不同的引物:包含靶区的两种单链互补物各一种。引物本身是单链序列,但比靶区的长度短得多,只能补充每个链3'端非常短的序列。
- 确定合适的退火温度至关重要,因为退火温度对影响效率和特异性有很大影响。该温度必须足够低以允许引物与链的杂交,但是足够高以使杂交具有特异性,即引物应该 仅仅与链中完全互补的部分结合,而不是其他地方。如果温度过低,引物可能结合不完全。如果太高,引物可能根本不会结合。典型的退火温度大约在引物 Tm以下3–5 °C。只有当引物序列与模板序列非常接近时,互补碱基之间才会形成稳定的氢键。在这一步骤中,聚合酶结合引物-模板杂交体,并开始DNA形成。
- 延伸/伸长:这一步的温度取决于所用的DNA聚合酶; Taq(水生栖热菌)聚合酶耐热DNA聚合酶的最佳活性温度约为75-80°C(167-176°F),尽管该酶通常使用72°C(162°F)的温度。在该步骤中,DNA聚合酶通过从反应混合物中添加在5’-至-3’方向上与模板互补的游离DNA链来合成与DNA模板链互补的新DNA链,在新生(伸长)DNA链的末端,将dNTPs的5'-磷酸基与3'-羟基缩合。延伸所需的精确时间取决于所使用的DNA聚合酶和要扩增的DNA目标区域的长度。根据经验,在最佳温度下,大多数DNA聚合酶每分钟聚合一千个碱基。在最佳条件下(即,如果没有限制底物或试剂的限制),在每个延伸/延伸步骤中,DNA靶序列的数量加倍。随着每一个连续的循环,原始模板链加上所有新产生的链成为下一轮延伸的模板链,导致特定DNA目标区域的指数(几何)扩增。
- 变性、退火和伸长的过程构成一个单一的循环。将DNA目标扩增到数百万个拷贝需要多个周期。用于计算给定循环次数后形成的DNA拷贝数的公式是2 n,其中 n是周期数。因此,30个循环的反应集产生2个 30,或1073741824原始双链DNA目标区域的拷贝。
- 最终伸长率:此单一步骤是可选的,但在70 - 74°C的温度(158 - 165°F)下执行(PCR中大多数聚合酶最佳活性所需的温度范围)在最后一个PCR循环后5-15分钟,以确保任何剩余的单链DNA被完全延长。
- 最终保持:最后一步将反应室冷却至4–15 °C (39–59 °F),并且可以用于PCR产物的短期储存。

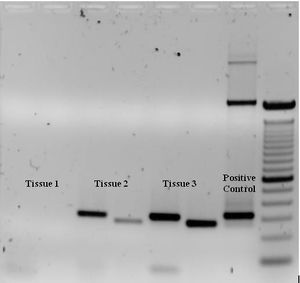
为了检查聚合酶链反应是否成功产生预期的目的DNA(有时也称为扩增子或扩增子),可以使用琼脂糖凝胶电泳进行聚合酶链反应产物的大小分离。聚合酶链反应产物的大小是通过与 DNA梯比较来确定的,DNA梯是一种分子量标记,包含与聚合酶链反应产物一起在凝胶上运行的已知大小的DNA片段。
1.2 阶段
与其他化学反应一样,聚合酶链反应的反应速率和效率受到限制因素的影响。因此,整个聚合酶链反应过程可以根据反应进程进一步分为三个阶段:
- 指数放大:在每个循环中,产物的量加倍(假设100%反应效率)。经过30个周期后,DNA的单个拷贝可以增加到10亿个拷贝。从某种意义上说,一条离散DNA链的复制是在试管中于受控条件下进行的。该反应非常敏感:只需要少量的DNA。
- 稳定阶段当DNA聚合酶失去活性时,以及DNA聚合酶和引物等试剂的消耗会导致他们变得更加有限,反应会变慢。
- 平台期:由于试剂和酶耗尽,不再有产品积累。
2 优化编辑
实际上,PCR可能由于各种原因失败,部分原因是它对污染的敏感性导致虚假DNA产物的扩增。因此,已经开发了许多技术和程序来优化聚合酶链反应条件。[7][8]外来DNA的污染通过实验室协议和程序来解决,这些协议和程序将PCR前混合物与潜在的DNA污染物分开。[9]这通常包括将聚合酶链式反应设置区域与聚合酶链式反应产品的分析或纯化区域空间分离,使用一次性塑料制品,以及彻底清洁反应设置之间的工作表面。引物设计技术对于提高PCR产物产量和避免假产物的形成非常重要,使用替代缓冲组分或聚合酶可以帮助扩增DNA的长区域或其他有问题的区域。在缓冲体系中加入试剂,如甲酰胺,可以提高聚合酶链反应的特异性和产率。[9]可以对理论聚合酶链反应结果进行计算机模拟(电子聚合酶链反应),以帮助引物设计。[10]
3 应用程序编辑
3.1 选择性脱氧核糖核酸分离
PCR允许从基因组DNA中通过选择性扩增DNA的特定区域分离DNA片段。聚合酶链反应的使用增加了许多方法,例如用于Southern或 Northern杂交的杂交探针和 DNA克隆,这需要更大量的DNA,代表特定的DNA区域。PCR为这些技术提供了大量的纯DNA,使得即使从非常少量的原始材料中也能够分析DNA样品。
聚合酶链反应的其他应用包括脱氧核糖核酸测序以确定未知的聚合酶链反应扩增序列,其中一个扩增引物可用于Sanger测序、分离脱氧核糖核酸序列以加速重组脱氧核糖核酸技术,该技术涉及将脱氧核糖核酸序列插入到质粒、噬菌体或粘粒(取决于大小)或另一生物体的遗传物质中。细菌菌落(如大肠杆菌)可以通过聚合酶链反应快速筛选正确的DNA 载体构建体。[11]聚合酶链反应也可用于遗传指纹;一种法医技术,用于通过不同的PCR方法比较实验性DNA来鉴定人或其他生物。
一些聚合酶链反应的“指纹”方法具有很高的辨别能力,可以用于识别个体之间的遗传关系,例如亲子关系或兄弟姐妹之间的遗传关系,并用于亲子鉴定(图4)。当使用特定的分子钟(即微生物的16S rRNA 和recA基因)时,该技术也可以用于确定生物体之间的进化关系。[12]

3.2 DNA的扩增和定量
因为PCR扩增了目的DNA区域,所以PCR可以用于分析极少量的样品。这对于法医检定法,当时只有微量的DNA可作为证据。聚合酶链反应也可用于分析古代DNA那已经有数万年的历史了。这些基于聚合酶链反应的技术已经成功地应用于动物身上,例如四万年前猛犸,以及人类DNA,应用范围从分析埃及木乃伊识别一个俄罗斯沙皇和英国国王理查三世遗体的鉴定。[13]
定量聚合酶链反应或者实时聚合酶链反应[14]不要与逆转录-聚合酶链反应方法混淆)允许估计样品中存在的给定序列的量——这种技术通常用于定量确定基因表达的水平。定量PCR是一种已建立的DNA定量工具,用于测量每轮PCR扩增后DNA产物的积累。
定量PCR允许实时定量和检测特定的DNA序列,因为它在合成过程中测量浓度。有两种方法可以同时检测和定量。第一种方法是使用在DNA双链之间非特异性保留的荧光染料。第二种方法涉及编码特定序列并被荧光标记的探针。只有在探针与其互补DNA杂交后,才能使用这些方法检测DNA。一种有趣的技术组合是实时PCR和逆转录。这种复杂的技术被称为RT-qPCR,允许定量少量RNA。通过这种组合技术,mRNA被转化为cDNA,并用qPCR进一步定量。这种技术降低了聚合酶链反应终点出错的可能性,[15]增加了检测与遗传疾病如癌症相关的基因的机会。[16]实验室使用RT-qPCR来灵敏地测量基因调控。
3.3 医疗和诊断应用
未来的父母可以被测试是否是遗传携带者,或者他们的孩子可以被测试是否真的受到了疾病的影响。[16]用于产前检测的DNA样品可以通过羊膜穿刺术、绒毛取样或甚至通过分析在母亲血流中循环的稀有胎儿细胞获得。聚合酶链反应分析对于植入前遗传诊断也是至关重要的,在植入前遗传诊断中,对发育中胚胎的单个细胞进行突变测试。
- 聚合酶链反应也可以用作以下敏感试验的一部分组织分型,对器官移植至关重要。截至2008年,甚至有人提议用聚合酶链反应检测取代传统的血型抗体检测。[16]
- 许多形式的癌症涉及致癌基因的改变。通过使用基于聚合酶链反应的测试来研究这些突变,治疗方案有时可以根据患者的不同而不同。聚合酶链反应可以早期诊断恶性疾病,如白血病和淋巴瘤,这是目前癌症研究中发展最快的,并且已经被常规使用。PCR检测可以直接在基因组DNA样品上进行,以检测易位特异性恶性细胞,其灵敏度至少是其他方法的10,000倍。[17]聚合酶链反应在医学领域非常有用,因为它允许分离和扩增肿瘤抑制剂。例如,定量PCR可以用于量化和分析单细胞,以及识别DNA、mRNA和蛋白质的确认和组合。[15]
3.4 传染病应用
聚合酶链反应可以快速和高度特异性地诊断传染疾病病,包括由细菌或病毒引起的疾病。[18]聚合酶链反应还允许从组织培养试验和动物模型中鉴定不可培养或生长缓慢的微生物,例如分枝杆菌、厌氧菌或病毒。聚合酶链反应在微生物学中诊断应用的基础是检测感染因子和借助特定基因从病原菌株中区分非病原性。[18][19]
通过PCR,传染病生物体的表征和检测已经在以下方面发生了革命性的变化:
- 这人类免疫缺陷病毒(或艾滋病毒),是一个很难找到和根除的目标。最早的感染测试依赖于血液中循环的病毒抗体的存在。然而,抗体直到感染后许多周才会出现,母体抗体掩盖了新生儿的感染,而对抗感染的治疗药物不影响抗体。PCR 测试已经被开发出来,它可以检测超过50,000个宿主细胞的DNA中的一个病毒基因组。[20]可以更早地检测感染,可以直接筛查捐赠的血液中的病毒,可以立即检测新生儿的感染,并且可以对抗病毒治疗的效果进行量化。
- 一些疾病生物,如肺结核,很难从病人身上取样,而且在实验室里生长缓慢。基于聚合酶链反应的检测可以方便地检测少量疾病生物(活的或死的)样品。详细的遗传分析也可以用来检测抗生素耐药性,从而允许立即有效的治疗。治疗效果也可以立即评估。
- 可通过PCR检测监测疾病病菌通过家畜或野生动物种群的传播。在许多情况下,可以检测和监测新毒性亚型的出现。也可以通过聚合酶链反应分析来确定导致早期流行的生物体的亚型。
- 病毒DNA可以通过PCR检测。所使用的引物必须对病毒DNA中的目标序列具有特异性,并且PCR可以用于病毒基因组的诊断分析或DNA测序。聚合酶链反应的高灵敏度允许在感染后不久甚至在疾病开始之前检测病毒。[18]这种早期发现可能会给医生治疗带来很长的准备时间。患者体内的病毒量(“病毒载量”)也可以通过基于聚合酶链反应的DNA定量技术进行定量(见下文)。
- 百日咳等疾病是由细菌百日咳杆菌引起的。这种细菌的特点是严重的急性呼吸道感染,影响到各种动物和人类,并导致许多幼儿死亡。百日咳毒素是一种蛋白质外毒素,通过两个二聚体与细胞受体结合,并与在细胞免疫中发挥作用的不同细胞类型如T淋巴细胞反应。[21]聚合酶链反应是一种重要的检测工具,可以检测百日咳毒素基因中的序列。这是因为聚合酶链反应对毒素有很高的灵敏度,并显示出快速的周转时间。与常规培养相比,聚合酶链反应对百日咳的诊断非常有效。[22]
3.5 法医应用
基于聚合酶链反应的遗传(或脱氧核糖核酸)指纹图谱协议的发展已在法医学中得到广泛应用:
- 遗传指纹以其最具辨别力的形式,可以从世界上所有人群中独一无二地区分任何一个人。微小的DNA样本可以从犯罪现场,和比较的从嫌疑人那里,或者从DNA资料库早期的证据或罪犯。这些测试的简单版本通常用于在刑事调查中快速排除嫌疑人。几十年前的犯罪证据可以被检验,确认或免责最初被定罪的人。
- 通过分析犯罪现场发现的证据,法医DNA分型已经成为鉴定犯罪嫌疑人或为其开脱的有效方法。人类基因组有许多重复区域,可以在基因序列中或基因组的非编码区域中发现。具体来说,高达40%的人类DNA是重复的。[16]基因组中这些重复的非编码区域有两种不同的类别。第一类称为可变数目串联重复序列(VNTR),长度为10-100个碱基对,第二类称为短串联重复序列(STR),由重复的2-10个碱基对部分组成。PCR用于使用位于每个重复区域侧翼的引物扩增几个众所周知的VNTRs和str。从任何个体获得的每个str片段的大小将表明存在哪些等位基因。通过分析一个人的几个str,会发现每个人的一组等位基因在统计上可能是唯一的。[16]研究人员已经确定了人类基因组的完整序列。该序列可以通过NCBI网站轻松访问,并用于许多实际应用中。例如,联邦调查局已经汇编了一组用于识别的DNA标记位点,这些位点被称为组合DNA索引系统(CODIS) DNA数据库。[16]使用该数据库可以使用统计分析来确定DNA样品匹配的概率。PCR是一种非常强大和重要的分析工具,用于法医DNA分型,因为研究人员只需要非常少量的目标DNA用于分析。例如,一根附着毛囊的人的头发有足够的DNA进行分析。类似地,一些精子、指甲下的皮肤样本或少量血液可以为结论性分析提供足够的DNA。[16]
- 脱氧核糖核酸指纹中较小的区别有助于亲子鉴定,在亲子鉴定中,一个人和他的近亲相匹配。可以测试未知人类遗骸的DNA,并与可能的父母、兄弟姐妹或儿童进行比较。类似的测试可以用来确认被收养(或绑架)孩子的亲生父母。新生儿的实际生父也可以被确认(或排除)。
- PCR AMGX/AMGY设计已被证明不仅有助于从非常少量的基因组中扩增DNA序列。然而,它也可以用于法医骨骼样本的实时性别确定。这为我们提供了一种强大而有效的方法,不仅可以确定古代标本的性别,还可以确定当前犯罪嫌疑人的性别。[23]
3.6 研究应用
PCR已经应用于分子遗传学的许多研究领域:
- 聚合酶链反应允许快速生产短DNA片段,即使已知的引物序列不超过两个。聚合酶链反应的这种能力增强了许多方法,例如生成杂交 探针用于 Southern 或northern blot 杂交。聚合酶链反应为这些技术提供了大量的纯DNA,有时是单链的,甚至可以从非常少量的起始材料中进行分析。
- 脱氧核糖核酸测序的任务也可以通过聚合酶链反应来辅助完成。已知的DNA片段可以很容易地从患有遗传疾病突变的患者体内产生。对扩增技术的修饰可以从完全未知的基因组中提取片段,或者可以仅产生感兴趣区域的单链。
- 聚合酶链反应有许多应用于传统的 DNA克隆。它可以从更大的基因组中提取片段以插入载体,这可能只有少量可用。使用一组“载体引物”,它还可以分析或提取已经插入载体的片段。聚合酶链反应方案的一些改变可以产生突变(通用的或定点的)插入片段。
- 序列标签站点是一个过程,其中PCR被用作基因组的特定片段存在于特定克隆中的指示物。人类基因组计划发现这一应用对于绘制他们测序的粘粒克隆以及协调不同实验室的结果至关重要。
- 聚合酶链反应的一个令人兴奋的应用是对来自远古来源的DNA进行系统进化分析,例如在尼安德特人的复原骨骼中、从猛犸的冷冻组织中或从埃及木乃伊的大脑中发现的DNA已经被放大和测序。[24]在某些情况下,这些来源的高度降解的DNA可能在扩增的早期阶段重新组装。
- 聚合酶链反应的一个常见应用是对以下基因表达模式的研究。组织(甚至单个细胞)可以在不同阶段进行分析,以了解哪些基因变得活跃,哪些基因被关闭。该应用还可以使用定量聚合酶链反应来定量表达的实际水平
- 聚合酶链反应同时扩增单个精子中几个基因座的能力[24]大大增强了极大地增强了通过研究减数分裂后染色体交叉来进行基因定位的传统任务。通过分析数千个单个精子,已经直接观察到非常紧密基因座之间罕见的交叉事件。类似地,可以分析异常的缺失、插入、易位或倒位,所有这些都无需等待(或支付)漫长而艰苦的受精、胚胎发生等过程。
- 定点突变:聚合酶链反应可用于产生突变基因,突变由科学家随意选择。可以选择这些突变来理解蛋白质是如何完成其功能的,并改变或改善蛋白质功能。
4 优势编辑
5 限制编辑
聚合酶链反应的一个主要限制是,为了产生允许其选择性扩增的引物,需要关于目标序列的先前信息。[26]这意味着,通常情况下,PCR用户必须知道两个单链模板中每个模板上目标区域上游的精确序列,以确保DNA聚合酶正确结合引物-模板杂交体,并随后在DNA合成过程中产生整个目标区域。
像所有酶一样,DNA聚合酶也容易出错,这反过来会导致产生的PCR片段发生突变。[27]
PCR的另一个限制是,即使是最少量的污染DNA也可以被扩增,导致误导或模糊的结果。为了最大限度地减少污染的可能性,调查人员应该为试剂制备、聚合酶链反应和产品分析预留单独的房间。试剂应分配到一次性的等分试样中。应经常使用带有一次性柱塞和超长移液器吸头的移液器。[28]
6 变化编辑
- 等位基因特异性聚合酶链反应:基于单核苷酸变异的诊断或克隆技术(snv不要与 SNPs 混淆)(患者的单碱基差异)。它需要事先知道DNA序列,包括等位基因之间的差异,并使用3’端包含SNV的引物(通常包含SNV周围的碱基对缓冲液)。在模板和引物不匹配的情况下,严格条件下的PCR扩增效率要低得多,因此用单核苷酸多态性特异性引物成功扩增表明序列中存在特异性单核苷酸多态性。[28]有关更多信息,请参见单核苷酸多态性基因分型。
- 装配聚合酶链反应或者聚合酶循环组件:通过对具有短重叠片段的长寡核苷酸池进行PCR来人工合成长DNA序列。寡核苷酸在有义和反义方向之间交替,重叠片段决定聚合酶链反应片段的顺序,从而选择性地产生最终的长DNA产物。[29]
- 不对称聚合酶链反应:优先扩增双链DNA模板中的一条DNA链。它用于测序和杂交探测,其中只需要扩增两条互补链中的一条。聚合酶链反应照常进行,但扩增目标链的引物过量。由于限制性引物用完后,反应后期的扩增速度较慢(算术),因此需要额外的PCR循环。[30]这个过程最近的一个修改,称为L²线性-A之后-T他-Exponential-PCR (LATE-PCR)使用比过量引物熔点更高的限制性引物( Tm ),以保持反应效率,因为限制性引物浓度在反应中期降低。[31]
- 对流聚合酶链反应:一种伪等温PCR方法。将溶液置于热梯度下,而不是反复加热和冷却聚合酶链反应混合物。由此产生的热不稳定性驱动对流流动自动将聚合酶链反应试剂从热区域和冷区域打乱,从而反复启动聚合酶链反应。[32]通过利用混沌流场的出现,可以优化热边界条件和PCR外壳的几何形状等参数,以产生特异和快速的PCR。[33]这种对流聚合酶链反应设置显著降低了设备功率需求和操作时间。
- 拨号聚合酶链反应:一种高度并行的方法,用于为基因合成检索精确的DNA分子。在大规模平行测序之前,用独特的侧翼标签修饰复杂的DNA分子库。然后,标签导向引物能够通过聚合酶链反应检索具有所需序列的分子。[34]
- 数字聚合酶链反应 (dPCR):用于测量DNA样品中目标DNA序列的数量。DNA样品被高度稀释,使得在并行运行许多PCR后,其中一些PCR不会接收到目标DNA的单个分子。目标DNA浓度是使用负结果的比例计算的。因此得名“数字聚合酶链反应”。
- Helicase-dependent amplification:与传统PCR相似,但使用恒温而不是通过变性和退火/延伸循环。 DNA解旋酶,一种解开DNA的酶,被用来代替热变性。[35]
- 热启动聚合酶链式反应:一种在PCR的初始建立阶段减少非特异性扩增的技术。它可以通过将反应组分加热到变性温度(例如95 c)在加入聚合酶之前。[36]已经开发了特殊的酶系统,通过结合抗体来抑制聚合酶在环境温度下的活性[37][37]或者通过共价键结合的抑制剂的存在,该抑制剂仅在高温活化步骤后解离。热启动/冷完成聚合酶链反应是通过新的杂合聚合酶实现的,这些酶在环境温度下不活跃,在伸长温度下立即激活。
- 电子聚合酶链反应(数字聚合酶链反应、虚拟聚合酶链反应、电子聚合酶链反应、电子聚合酶链反应)是指计算工具使用给定的一组引物 ( 探针)来扩增来自测序的基因组或转录组的DNA 序列,用于计算理论聚合酶链反应结果。电子PCR被提出作为分子生物学的教育工具。[38]
- 序列间特异性聚合酶链反应(ISSR):一种用于DNA指纹识别的聚合酶链反应方法,它放大简单重复序列之间的区域,以产生扩增片段长度的独特指纹。[39]
- 反向聚合酶链反应:通常用于识别基因组插入片段周围的侧翼序列。它涉及一系列的脱氧核糖核酸消化和自连接,在未知序列的两端产生已知序列。[40]
- 连接介导的聚合酶链反应:使用连接到感兴趣的DNA的小DNA接头和退火到DNA接头的多个引物;它已被用于 DNA测序、基因组行走和 DNA足迹法。[41]
- 甲基化特异性聚合酶链反应(MSP):由史蒂芬·贝林和詹姆斯·赫尔曼在约翰·霍普金斯大学医学院开发,[42]用于检测基因组DNA中CpG岛的甲基化。DNA首先用亚硫酸氢钠处理,亚硫酸氢钠将未甲基化的胞嘧啶碱基转化为尿嘧啶,尿嘧啶被PCR引物识别为胸腺嘧啶。然后在修饰的DNA上进行两次PCR,使用除引物序列中任何CpG岛以外相同的引物组。在这些点上,一组引物用胞嘧啶识别DNA以扩增甲基化的DNA,一组引物用尿嘧啶或胸腺嘧啶识别DNA以扩增未甲基化的DNA。使用qPCR进行MSP也可以获得甲基化的定量而不是定性信息。
- 微型引物聚合酶链反应:使用耐热聚合酶(S-Tbr),可以从短引物延伸到9或10个核苷酸。这种方法允许PCR靶向较小的引物结合区域,并用于扩增保守的DNA序列,如16S(或真核18S) rRNA基因。[43]
- 多重连接依赖探针扩增(MLPA):允许用单个引物对扩增多个靶标,从而避免多重PCR的分辨率限制(见下文)。
- 多重聚合酶链反应:由单个PCR混合物中的多个引物组组成,以产生对不同DNA序列特异的不同大小的扩增子。通过同时靶向多个基因,可以从单次测试中获得额外的信息,否则将需要几次试剂和更多时间来执行。每个引物组的退火温度必须优化,以在单个反应中正确工作,并符合扩增子尺寸。也就是说,当通过凝胶电泳可视化时,它们的碱基对长度应该足够不同以形成不同的条带。
- 纳米粒子辅助聚合酶链反应:一些纳米粒子(NPs)可以提高PCR的效率(因此被称为纳米PCR),一些甚至可以优于原始的PCR增强剂。据报道,量子点可以提高聚合酶链反应的特异性和效率。单壁碳纳米管和多壁碳纳米管能有效增强长聚合酶链反应的扩增。纳米碳粉(成交堆积)可以提高重复聚合酶链反应和长聚合酶链反应的效率氧化锌,二氧化钛发现银纳米粒子能提高聚合酶链反应的产率。以前的数据表明非金属NP保留了可接受的放大保真度。鉴于许多纳米粒子能够提高聚合酶链反应效率,很明显纳米粒子技术的改进和产品开发有很大的潜力。[44][45]
- 嵌套聚合酶链反应:通过减少DNA非特异性扩增的背景,提高DNA扩增的特异性。两组引物用于两个连续的PCR。在第一个反应中,一对引物用于产生DNA产物,除了预期的靶之外,该产物可能仍然由非特异性扩增的DNA片段组成。然后用一组引物将产物用于第二次聚合酶链反应,所述引物的结合位点完全或部分不同于第一次反应中使用的每个引物,并且位于其中的3’。嵌套式PCR在特异性扩增长DNA片段方面通常比传统PCR更成功,但它需要更详细的目标序列知识。
- 重叠延伸聚合酶链反应或者通过重叠延伸拼接(SOEing):一种用于将两个或多个含有互补序列的DNA片段拼接在一起的基因工程技术。它用于连接含有基因、调节序列或突变的DNA片段;这项技术可以创造特定的长DNA构建体。它还可以将缺失、插入或点突变引入DNA序列。[46][47]
- PAN-AC:使用等温条件进行扩增,并可用于活细胞。[48][49]
- 定量聚合酶链反应(qPCR):用于测量目标序列的数量(通常是实时的)。它定量测量DNA、cDNA或RNA的起始量。定量PCR 通常用于确定样品中是否存在DNA序列及其在样品中的拷贝数。定量聚合酶链反应具有非常高的精确度。定量PCR方法使用荧光染料,如Sybr Green、EvaGreen或含荧光团的DNA探针,如TaqMan ,实时测量扩增产物的量。它有时也被缩写为逆转录-聚合酶链反应(实时的聚合酶链反应),但这个缩写只能用于 逆转录PCR 。定量PCR是定量PCR 的合适缩写。
- 逆转录聚合酶链反应(逆转录-聚合酶链反应):用于从RNA中扩增DNA。逆转录酶将核糖核酸逆转录成cDNA ,然后通过聚合酶链反应进行扩增。逆转录-聚合酶链反应广泛用于表达谱,以确定基因的表达或鉴定RNA转录物的序列,包括转录起始和终止位点。如果基因的基因组DNA序列是已知的,逆转录-聚合酶链反应可以用来绘制基因中外显子和内含子的位置。基因的5’端(对应于转录起始位点)通常通过 RACE-PCR (快速扩增cDNA末端)中。
- 核糖核酸酶H依赖性聚合酶链反应(rhPCR):PCR的一种改进,利用带有3’延伸区的引物,该延伸区可以被耐热核糖核酸酶HII酶去除。该系统减少了引物二聚体,并允许用更高数量的引物进行多重反应。[50]
- 单一特异性引物聚合酶链反应(SSP-PCR):即使序列信息仅在一端可用,也允许双链DNA的扩增。这种方法允许扩增只有部分序列信息可用的基因,并允许单向基因组从染色体的已知区域步行到未知区域。[51]
- 固相聚合酶链反应:包含多种含义,包括 Polony Amplification (例如,聚合酶链反应菌落来源于凝胶基质),桥聚合酶链反应[52](引物共价连接到固相支持物表面)、常规固相聚合酶链反应(其中不对称聚合酶链反应在带有固相支持物的引物存在下应用,所述引物的序列与其中一个水性引物匹配)和增强型固相聚合酶链反应[53](其中传统的固相聚合酶链反应可以通过使用高Tm和嵌套的固相支持物引物并任选应用热“步骤”来促进固相支持物引发来改进)。
- 自杀聚合酶链反应:通常用于古遗传学或其他研究中,其中避免假阳性并确保扩增片段的特异性是最高优先级。最初是在一项研究中描述的,该研究旨在验证从14世纪的坟墓中获得的牙科样本中是否存在微生物鼠疫耶尔森菌,据说这些坟墓是在中世纪的黑死病流行期间被鼠疫杀死的。[54]该方法规定在聚合酶链反应中只使用一次任何引物组合(因此称为“自杀”),这种组合不应用于任何阳性对照聚合酶链反应,引物应始终针对以前在实验室中从未使用该引物或任何其他引物组扩增的基因组区域。这确保了实验室中不存在来自先前PCR反应的污染DNA,否则这些DNA可能会产生假阳性。
- 热不对称交错聚合酶链反应:用于分离已知序列侧翼的未知序列。在已知序列中,TAIL-PCR使用一对具有不同退火温度的嵌套引物;简并引物用于从未知序列向另一个方向扩增。[55]
- 触地聚合酶链反应(逐步下降聚合酶链反应):PCR的一种变体,旨在通过随着PCR循环的进行逐渐降低退火温度来减少非特异性背景。初始循环的退火温度通常为几度(3-5 c)高于Tm在使用的引物中,在后期的循环中,它是几度(3-5 c)低于底漆Tm。较高的温度赋予引物结合更大的特异性,较低的温度允许从初始循环中形成的特定产物中进行更有效的扩增。[56]
- 万能快走:对于基因组行走和遗传指纹识别,使用比传统的“单侧”方法更特异性的“双侧”PCR(仅使用一个基因特异性引物和一个通用引物——这可能导致动脉事实上的“噪音”)[57]这是由于套索结构形成的机制。UFW的流线型衍生物是LaNe RAGE(用于快速扩增基因组DNA末端的套索依赖性巢式PCR),[58]' 5'RACE LaNe[59]和3'RACE LaNe。[60]
7 历史编辑
1971年的一篇论文《分子生物学杂志》作者:Kjell Kleppe (挪威语)高宾德·科兰纳实验室的同事首先描述了一种在试管内使用酶分析法用引物复制短DNA模板的方法。[61]然而,这种基本聚合酶链反应原理的早期表现在当时并没有受到太多关注,聚合酶链式反应在1983年的发明通常归功于凯利·穆利斯。[62]
当穆利斯在1983年开发聚合酶链反应时,他在加利福尼亚州的 Emeryville 为塞特斯公司工作,该公司是首批生物技术公司之一,他负责合成DNA短链。穆利斯写道,有一天晚上,他开车沿着太平洋海岸高速公路巡航时,第一次想到了聚合酶链反应的想法。[63]当他意识到他已经发明了一种通过由DNA聚合酶驱动的重复复制周期来扩增任何DNA区域的方法时,他正在用一种新的分析DNA变化(突变)方法。在科学美国人穆利斯总结道:“从遗传物质DNA的一个分子开始,聚合酶链反应可以在一个下午产生1000亿个类似的分子。这个反应很容易执行。它只需要一个试管、一些简单的试剂和一个热源。”[64]1988年,DNA指纹首次用于亲子鉴定。[65]
穆利斯因其发明于1993年被授予诺贝尔化学奖,七年前他和他在塞特斯的同事首次将他的建议付诸实践。[66]mullis 1985年与R. K .斋祀和H. A. Erlich合著的论文《β-珠蛋白基因组序列的酶促扩增和用于镰刀形红细胞贫血症诊断的限制性位点分析》——聚合酶链式反应发明(PCR)——荣获2017年美国化学学会历史分期化学奖化学突破奖。[66][67]
关于其他科学家对穆利斯工作的智力和实践贡献,以及他是否是PCR原理的唯一发明者,仍然存在一些争议(见下文)。
聚合酶链反应方法的核心是使用合适的能够耐受高温的脱氧核糖核酸聚合酶90 °C (194 °F)在每一个复制循环后,在 DNA双螺旋中分离两条DNA链所必需的。最初用于体外实验的DNA聚合酶预示着PCR不能耐受这些高温。[68]因此,早期的DNA复制过程非常低效和耗时,并且需要大量的DNA聚合酶和在整个过程中的连续处理。
1976年发现了 Taq聚合酶——一种从嗜热细菌中纯化的DNA聚合酶,水生栖热菌,它自然地生活在诸如[68]温泉之类的高温(50至80°C(122至176°F))环境中,为PCR方法的显著改进铺平了道路。从T.aquaticus分离出的DNA聚合酶在高温下稳定,即使在DNA变性之后仍保持活性,[68]因此不需要在每个循环后添加新的DNA聚合酶。[68]这允许基于自动热循环仪的DNA扩增过程。
7.1 专利纠纷
聚合酶链反应技术由凯利·穆利斯获得专利,并转让给塞特斯公司,穆利斯1983年发明这项技术时就在该公司工作。Taq聚合酶也被专利所保护。已经有几起与该技术相关的高调诉讼,包括杜邦提起的一起不成功的诉讼。制药公司罗氏在1992年购买了这些专利的权利,目前拥有那些仍然受到保护的权利。
罗氏和Promega 之间围绕Taq聚合酶酶的相关专利战仍在世界各地的几个管辖区进行。法律争论已经超出了2005年3月28日到期的原始PCR和Taq聚合酶专利的寿命。[68]
参考文献
- [1]
^Cheng, S.; Fockler, C.; Barnes, W. M.; Higuchi, R. (1994). "Effective Amplification of Long Targets from Cloned Inserts and Human Genomic DNA". Proceedings of the National Academy of Sciences. 91 (12): 5695–5699. Bibcode:1994PNAS...91.5695C. doi:10.1073/pnas.91.12.5695. PMC 44063. PMID 8202550..
- [2]
^Carr AC, Moore SD (2012). Lucia A, ed. "Robust quantification of polymerase chain reactions using global fitting". PLoS ONE. 7 (5): e37640. Bibcode:2012PLoSO...737640C. doi:10.1371/journal.pone.0037640. PMC 3365123. PMID 22701526..
- [3]
^"Polymerase Chain Reaction (PCR)". National Center for Biotechnology Information, U.S. National Library of Medicine..
- [4]
^"PCR". Genetic Science Learning Center, University of Utah..
- [5]
^Pavlov, A. R.; Pavlova, N. V.; Kozyavkin, S. A.; Slesarev, A. I. (2004). "Recent developments in the optimization of thermostable DNA polymerases for efficient applications☆". Trends in Biotechnology. 22 (5): 253–260. doi:10.1016/j.tibtech.2004.02.011. PMID 15109812..
- [6]
^Rychlik W, Spencer WJ, Rhoads RE (1990). "Optimization of the annealing temperature for DNA amplification in vitro". Nucleic Acids Res. 18 (21): 6409–6412. doi:10.1093/nar/18.21.6409. PMC 332522. PMID 2243783..
- [7]
^Borman, Jon; Schuster, David; Li, Wu-bo; Jessee, Joel; Rashtchian, Ayoub (2000). "PCR from problematic templates" (PDF). Focus. 22 (1): 10..
- [8]
^Bogetto, Prachi and Waidne, Lisa (2000). "Helpful tips for PCR" (PDF). Focus. 22 (1): 12.CS1 maint: Multiple names: authors list (link).
- [9]
^Joseph Sambrook & David W. Russel (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 978-0-879-69576-7.第八章:聚合酶链反应体外扩增脱氧核糖核酸.
- [10]
^"Electronic PCR". NCBI – National Center for Biotechnology Information. Retrieved 13 March 2012..
- [11]
^Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). "Thermostable DNA Polymerases for a Wide Spectrum of Applications: Comparison of a Robust Hybrid TopoTaq to other enzymes". In Kieleczawa J. DNA Sequencing II: Optimizing Preparation and Cleanup. Jones and Bartlett. pp. 241–257. ISBN 978-0-7637-3383-4..
- [12]
^Pombert JF, Sistek V, Boissinot M, Frenette M (2009). "Evolutionary relationships among salivarius streptococci as inferred from multilocus phylogenies based on 16S rRNA-encoding, recA, secA, and secY gene sequences". BMC Microbiol. 9: 232. doi:10.1186/1471-2180-9-232. PMC 2777182. PMID 19878555..
- [13]
^"Chemical Synthesis, Sequencing, and Amplification of DNA (class notes on MBB/BIO 343)". Arizona State University. Archived from the original on 9 October 1997. Retrieved 29 October 2007..
- [14]
^Bustin, S. A.; Benes, V.; Garson, J. A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M. W.; Shipley, G. L.; Vandesompele, J.; Wittwer, C. T. (2009). "The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments". Clinical Chemistry. 55 (4): 611–622. doi:10.1373/clinchem.2008.112797. PMID 19246619..
- [15]
^Garibyan, Avashia (March 2013). "Polymerase Chain Reaction". Journal of Investigative Dermatology. 133 (3): 1–4. doi:10.1038/jid.2013.1. PMC 4102308. PMID 23399825..
- [16]
^Ninfa, Alexander; Ballou, David; Benore, Marilee (2009). Fundamental Laboratory Approaches for Biochemistry and Biotechnology. United States: Wiley. pp. 408–410. ISBN 978-0470087664..
- [17]
^Tomar, Rukam (2010). Molecular Markers and Plant Biotechnology. Pitman Pura, New Delhi: New India Publishing Agency. p. 188. ISBN 978-93-80235-25-7..
- [18]
^Cai, H; Caswell JL; Prescott JF (March 2014). "Nonculture Molecular Techniques for Diagnosis of Bacterial Disease in Animals: A Diagnostic Laboratory Perspective". Veterinary Pathology. 51 (2): 341–350. doi:10.1177/0300985813511132. PMID 24569613..
- [19]
^Salis AD (2009). "Applications in Clinical Microbiology". Real-Time PCR: Current Technology and Applications. Caister Academic Press. ISBN 978-1-904455-39-4..
- [20]
^Kwok, S.; Mack, D. H.; Mullis, K. B.; Poiesz, B.; Ehrlich, G.; Blair, D.; Friedman-Kien, A.; Sninsky, J. J. (1987). "Identification of human immunodeficiency virus sequences by using in vitro enzymatic amplification and oligomer cleavage detection". Journal of Virology. 61 (5): 1690–4. PMC 254157. PMID 2437321..
- [21]
^Finger, Horst; von Koenig, Carl Heinz Wirsing (1996). Baron, Samuel, ed. Medical Microbiology (4th ed.). Galveston (TX): University of Texas Medical Branch at Galveston. ISBN 978-0963117212. PMID 21413270..
- [22]
^Yeh, Sylvia H.; Mink, ChrisAnna M. (2012). "Netter's Infectious Diseases". Netter's Infectious Diseases: 11–14. doi:10.1016/B978-1-4377-0126-5.00003-3..
- [23]
^Alonso, A (2004-01-28). "Real-time PCR designs to estimate nuclear and mitochondrial DNA copy number in forensic and ancient DNA studies". Forensic Science International. 139 (2–3): 141–149. doi:10.1016/j.forsciint.2003.10.008. PMID 15040907..
- [24]
^Boehnke, M.; Arnheim, N.; Li, H.; Collins, F. S. (1989). "Fine-structure genetic mapping of human chromosomes using the polymerase chain reaction on single sperm: Experimental design considerations". American Journal of Human Genetics. 45 (1): 21–32. PMC 1683385. PMID 2568090..
- [25]
^Garibyan, Lilit; Avashia, Nidhi (2013-03-01). "Polymerase Chain Reaction". Journal of Investigative Dermatology. 133 (3): 1–4. doi:10.1038/jid.2013.1. PMC 4102308. PMID 23399825..
- [26]
^Garibyan L, Avashia N (2013). "Polymerase Chain Reaction". Journal of Investigative Dermatology. 133 (3): 1–4. doi:10.1038/jid.2013.1. PMC 4102308. PMID 23399825..
- [27]
^Zhou, Y H; Zhang, X P; Ebright, R H (1991-11-11). "Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase". Nucleic Acids Research. 19 (21): 6052. doi:10.1093/nar/19.21.6052. PMC 329070. PMID 1658751..
- [28]
^Schochetman, Gerald; Ou, Chin-Yih; Jones, Wanda K. (1988). "Polymerase Chain Reaction". The Journal of Infectious Diseases. 158 (6): 1154–1157. doi:10.1093/infdis/158.6.1154. JSTOR 30137034..
- [29]
^Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995). "Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides". Gene. 164 (1): 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320..
- [30]
^Innis MA, Myambo KB, Gelfand DH, Brow MA (1988). "DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA". Proc Natl Acad Sci USA. 85 (24): 9436–9440. Bibcode:1988PNAS...85.9436I. doi:10.1073/pnas.85.24.9436. PMC 282767. PMID 3200828..
- [31]
^Pierce KE & Wangh LJ (2007). Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells. Methods in Molecular Medicine. 132. pp. 65–85. doi:10.1007/978-1-59745-298-4_7. ISBN 978-1-58829-578-1. PMID 17876077..
- [32]
^Krishnan, Madhavi; Ugaz, Victor; Burns, Mark (2002). "PCR in a Rayleigh-Benard convection cell". Science. 298 (5594): 793. doi:10.1126/science.298.5594.793. PMID 12399582..
- [33]
^Priye, Aashish; Hassan, Yassin; Ugaz, Victor (2013). "Microscale chaotic advection enables robust convective DNA replication". Analytical Chemistry. 85 (21): 10536–10541. doi:10.1021/ac402611s. PMID 24083802..
- [34]
^Schwartz JJ, Lee C, Shendure J (2012). "Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules". Nature Methods. 9 (9): 913–915. doi:10.1038/nmeth.2137. PMC 3433648. PMID 22886093..
- [35]
^Vincent M, Xu Y, Kong H (2004). "Helicase-dependent isothermal DNA amplification". EMBO Reports. 5 (8): 795–800. doi:10.1038/sj.embor.7400200. PMC 1249482. PMID 15247927..
- [36]
^Chou Q, Russell M, Birch DE, Raymond J, Bloch W (1992). "Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications". Nucleic Acids Research. 20 (7): 1717–1723. doi:10.1093/nar/20.7.1717. PMC 312262. PMID 1579465..
- [37]
^Sharkey, D. J.; Scalice, E. R.; Christy, K. G.; Atwood, S. M.; Daiss, J. L. (1994). "Antibodies as Thermolabile Switches: High Temperature Triggering for the Polymerase Chain Reaction". Bio/Technology. 12 (5): 506–509. doi:10.1038/nbt0594-506..
- [38]
^San Millan RM, Martinez-Ballesteros I, Rementeria A, Garaizar J, Bikandi J (2013). "Online exercise for the design and simulation of PCR and PCR-RFLP experiments". BMC Research Notes. 6: 513. doi:10.1186/1756-0500-6-513. PMC 4029544. PMID 24314313..
- [39]
^Zietkiewicz, E.; Rafalski, A.; Labuda, D. (1994). "Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification". Genomics. 20 (2): 176–83. doi:10.1006/geno.1994.1151. PMID 8020964..
- [40]
^Ochman H, Gerber AS, Hartl DL (1988). "Genetic Applications of an Inverse Polymerase Chain Reaction". Genetics. 120 (3): 621–623. PMC 1203539. PMID 2852134..
- [41]
^Mueller PR, Wold B (1988). "In vivo footprinting of a muscle specific enhancer by ligation mediated PCR". Science. 246 (4931): 780–786. doi:10.1126/science.2814500. PMID 2814500..
- [42]
^Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996). "Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands". Proc Natl Acad Sci USA. 93 (13): 9821–9826. Bibcode:1996PNAS...93.9821H. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415..
- [43]
^Isenbarger TA, Finney M, Ríos-Velázquez C, Handelsman J, Ruvkun G (2008). "Miniprimer PCR, a New Lens for Viewing the Microbial World". Applied and Environmental Microbiology. 74 (3): 840–9. doi:10.1128/AEM.01933-07. PMC 2227730. PMID 18083877..
- [44]
^Cenchao Shen; Wenjuan Yang; Qiaoli Ji; Hisaji Maki; Anjie Dong; Zhizhou Zhang (2009). "NanoPCR observation: different levels of DNA replication fidelity in nanoparticle-enhanced polymerase chain reactions". Nanotechnology. 20 (45): 455103. Bibcode:2009Nanot..20S5103S. doi:10.1088/0957-4484/20/45/455103. PMID 19822925..
- [45]
^Shen, Cenchao (2013). "An Overview of Nanoparticle-Assisted Polymerase Chain Reaction Technology". An Overview of Nanoparticle‐Assisted Polymerase Chain Reaction Technology. US: Wiley-Blackwell Publishing Ltd. pp. 97–106. doi:10.1002/9781118451915.ch5. ISBN 9781118451915..
- [46]
^Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989). "Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap exten-sion". Gene. 77 (1): 61–68. doi:10.1016/0378-1119(89)90359-4. PMID 2744488..
- [47]
^Moller, Simon (2006). PCR: The Basics. US: Taylor & Francis Group. p. 144. ISBN 9780415355476..
- [48]
^David F, Turlotte E (1998). "Une méthode d'amplification génique isotherme" [An Isothermal Amplification Method]. Comptes Rendus de l'Académie des Sciences, Série III. 321 (11): 909–914. Bibcode:1998CRASG.321..909D. doi:10.1016/S0764-4469(99)80005-5..
- [49]
^Fabrice David (September–October 2002). "Utiliser les propriétés topologiques de l'ADN: une nouvelle arme contre les agents pathogènes" (PDF). Fusion. Archived from the original (PDF) on 2007-11-28.(法语).
- [50]
^Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, Walder JA (August 2011). "RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers". BMC Biotechnology. 11: 80. doi:10.1186/1472-6750-11-80. PMC 3224242. PMID 21831278..
- [51]
^Shyamala, V.; Ferro-Luzzi, Ames G. (1993). Single Specific Primer-Polymerase Chain Reaction (SSP-PCR) and Genome Walking. Methods in Molecular Biology. 15. pp. 339–48. doi:10.1385/0-89603-244-2:339. ISBN 978-0-89603-244-6. PMID 21400290..
- [52]
^Bing DH, Boles C, Rehman FN, Audeh M, Belmarsh M, Kelley B, Adams CP (1996). "Bridge amplification: a solid phase PCR system for the amplification and detection of allelic differences in single copy genes". Genetic Identity Conference Proceedings, Seventh International Symposium on Human Identification. Archived from the original on 7 May 2001..
- [53]
^Khan Z, Poetter K, Park DJ (2008). "Enhanced solid phase PCR: mechanisms to increase priming by solid support primers". Analytical Biochemistry. 375 (2): 391–393. doi:10.1016/j.ab.2008.01.021. PMID 18267099..
- [54]
^Raoult, D; G Aboudharam; E Crubezy; G Larrouy; B Ludes; M Drancourt (2000-11-07). "Molecular identification by "suicide PCR" of Yersinia pestis as the agent of medieval black death". Proc. Natl. Acad. Sci. U.S.A. 97 (23): 12800–12803. Bibcode:2000PNAS...9712800R. doi:10.1073/pnas.220225197. PMC 18844. PMID 11058154..
- [55]
^Y.G. Liu & R. F. Whittier (1995). "Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking". Genomics. 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J. PMID 7759102..
- [56]
^Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991). "'Touchdown' PCR to circumvent spurious priming during gene amplification". Nucleic Acids Res. 19 (14): 4008. doi:10.1093/nar/19.14.4008. PMC 328507. PMID 1861999..
- [57]
^Myrick KV, Gelbart WM (2002). "Universal Fast Walking for direct and versatile determination of flanking sequence". Gene. 284 (1–2): 125–131. doi:10.1016/S0378-1119(02)00384-0. PMID 11891053..
- [58]
^"Full Text – LaNe RAGE: a new tool for genomic DNA flanking sequence determination"..
- [59]
^Park DJ (2005). "A new 5' terminal murine GAPDH exon identified using 5'RACE LaNe". Molecular Biotechnology. 29 (1): 39–46. doi:10.1385/MB:29:1:39. PMID 15668518..
- [60]
^Park DJ (2004). "3'RACE LaNe: a simple and rapid fully nested PCR method to determine 3'-terminal cDNA sequence". BioTechniques. 36 (4): 586–588, 590. doi:10.2144/04364BM04. PMID 15088375..
- [61]
^Kleppe K, Ohtsuka E, Kleppe R, Molineux I, Khorana HG (1971). "Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA's as catalyzed by DNA polymerases". J. Mol. Biol. 56 (2): 341–361. doi:10.1016/0022-2836(71)90469-4. PMID 4927950..
- [62]
^Rabinow, Paul (1996). Making PCR: A Story of Biotechnology. Chicago: University of Chicago Press. ISBN 978-0-226-70146-2..
- [63]
^Mullis, Kary (1998). Dancing Naked in the Mind Field. New York: Pantheon Books. ISBN 978-0-679-44255-4..
- [64]
^Mullis, Kary (1990). "The unusual origin of the polymerase chain reaction". Scientific American. 262 (4): 56–61, 64–5. Bibcode:1990SciAm.262d..56M. doi:10.1038/scientificamerican0490-56. PMID 2315679..
- [65]
^Patidar, Madhvika; Agrawal, Suraksha; Parveen, Farah; Khare, Parul (2015). "Molecular insights of saliva in solving paternity dispute". Journal of Forensic Dental Sciences. 7 (1): 76–79. doi:10.4103/0975-1475.150325. PMC 4330625. PMID 25709326..
- [66]
^"Kary B. Mullis – Nobel Lecture: The Polymerase Chain Reaction"..
- [67]
^Saiki, R.; Scharf, S; Faloona, F; Mullis, K.; Horn, G.; Erlich, H.; Arnheim, N (1985). "Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia". Science. 230 (4732): 1350–1354. doi:10.1126/science.2999980..
- [68]
^Saiki, R.; Scharf, S.; Faloona, F.; Mullis, K.; Horn, G.; Erlich, H.; Arnheim, N. (1985). "Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia". Science. 230 (4732): 1350–1354. doi:10.1126/science.2999980. PMID 2999980..
暂无