染料测序
编辑Illumina染料测序是一种用于测定脱氧核糖核酸中一连串碱基对的技术,也称为脱氧核糖核酸(DNA)测序。可逆终止化学概念由巴黎巴斯德研究所的布鲁诺·卡纳德(Bruno Canard)和西蒙·萨法蒂(Simon Sarfati)提出。[1][2]后来剑桥大学的尚卡尔·巴拉苏布拉马尼安(Shankar Balasubramanian)和大卫·克莱恩曼(David Klenerman)开发了这一技术,[3]他们随后创建了Solexa公司,后来被Illumina收购。这种测序方法基于可逆的染料--终止技术,当它们被引入到脱氧核糖核酸链中时,能够识别单个碱基。它也可用于全基因组和局部测序、转录组分析、宏基因组学、小核糖核酸发现、甲基化谱和全基因组蛋白质-核酸互作分析。[4]
1 概述编辑
Illumina测序技术分为三个基本步骤:扩增、测序和分析。这个过程从纯化的脱氧核糖核酸开始。脱氧核糖核酸被破碎成较小的片段,并加上接头、索引和其他种类的分子修饰——在扩增、测序和分析过程中作为参考点。经过修饰的脱氧核糖核酸被加入到一个专门的芯片上,进行扩增和测序。芯片底部有成千上万的寡核苷酸(人工合成的短脱氧核糖核酸片段)。它们被锚定在芯片上,能够抓取具有互补序列的脱氧核糖核酸片段。一旦片段被连接到芯片上,簇生成阶段就开始了。这一步产生大约一千份脱氧核糖核酸片段的拷贝。接下来,引物和修饰的核苷酸进入芯片。这些核苷酸具有可逆的3’阻断基团,迫使聚合酶一次只能添加一个核苷酸,同时这个核苷酸还有荧光标记。在每一轮合成之后,照相机会给芯片拍一张照片。计算机根据荧光标签的波长确定添加了什么样的碱基,并记录下来芯片上的每个点的位置。每轮反应后,未结合的分子被洗去。用化学剂去封闭步骤,去除(单个碱基)3’末端的封闭基团和染料。这个过程一直持续到完整的脱氧核糖核酸分子被测序。[5]利用这项技术,通过大规模并行测序,基因组中的成千上万个位置同时被测序。
2 程序编辑
2.1 标签化
脱氧核糖核酸纯化后的第一步是加上标签。转座酶随机将脱氧核糖核酸切割成小段(“标签”)。接头被添加在切割点的两侧(连接)。未连接接头的DNA链被洗掉。[5]
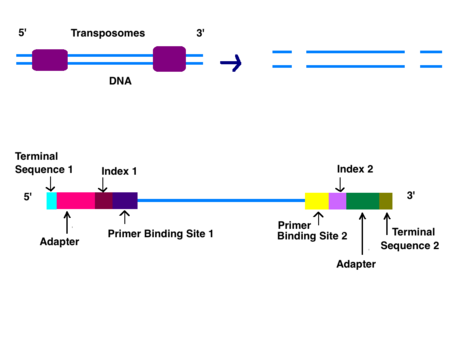
2.2 低循环扩增
下一步被称为低循环扩增。此步骤 (在DNA 片段上)添加引物结合序列、索引和末端序列。索引序列的长度通常是六个碱基对,在脱氧核糖核酸序列分析中用来鉴定样品。索引允许多达96个不同的样本一起进行扩增。在分析过程中,计算机会将所有具有相同索引的读取结果组合在一起。[6][7]末端序列用于将脱氧核糖核酸链连接到流动池上。Illumina采用“边合成边测序”方法。[7]这一过程发生在丙烯酰胺涂层的玻璃流动池内。[8]在流动池的底部覆盖有寡核苷酸 (短核苷酸序列),它们在测序过程中用于将脱氧核糖核酸链固定到适当位置。寡核苷酸与低循环扩增过程中添加到脱氧核糖核酸中的两种末端序列相匹配。当脱氧核糖核酸进入流动池内,其中一个接头连接到与其互补的寡核苷酸上。
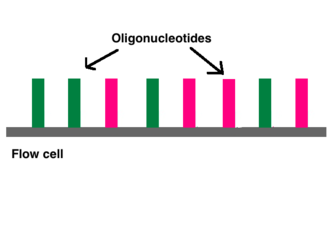
2.3 桥式扩增
一旦与流动池上寡核苷酸连接,簇生成就可以开始了。目的是创造许许多多相同的脱氧核糖核酸链。有些是正向链;其余的,是反向链。簇是通过桥式扩增产生的。聚合酶沿着一条脱氧核糖核酸链移动,形成互补链。原来的模板链被洗去,只留下反向链。在反向链的顶部有接头序列。脱氧核糖核酸链弯曲并与顶部接头序列互补的寡核苷酸杂交结合。聚合酶附着在反向链上,形成它的互补链(即与模板链相同)。此时的双链脱氧核糖核酸变性解链,每条链单独锚定在流动池上的寡核苷酸序列上。一个是反向链;另一个是先前的链。这一过程被称为桥式扩增,它同时发生在整个流动池的数千个簇中。
2.4 克隆扩增
脱氧核糖核酸链一次又一次进行弯曲与寡核苷酸互补结合。聚合酶将合成一条新的链来产生双链片段,然后该片段变性,使得一个区域中的所有脱氧核糖核酸链来自同一个DNA分子(克隆扩增)。克隆扩增对于质量控制非常重要。如果发现一条链有一个奇数序列,那么科学家可以检查反向链,以确保它有相同奇数的互补序列。正向和反向链起到检查防止假象的作用。因为Illumina测序使用的聚合酶,已发现过有碱基错误替换(的现象),[9]特别是在3’末端。[10]配对的末端读取结果结合簇生成可以确认错误发生(情况)。反向和正向链应该彼此互补,所有反向的读取结果应该相互匹配,所有正向读取结果也应该相互匹配。如果一条reads与其对应副本(通过它进行克隆)不够相似,则可能发生了错误。在一些实验室的分析中使用97%相似度作为最小阈值。[10]
2.5 边合成边测序
克隆扩增结束时,所有反向链都从流动池中洗掉,只留下正向链。引物结合到正向链上,聚合酶将荧光标记的核苷酸添加到脱氧核糖核酸链上。每轮只增加一个碱基。每一个核苷酸上都有一个可逆的终止剂,以防止一轮中多次添加核苷酸。使用四色化学发光,四个碱基每一个都有独特的荧光信号释放,每一轮之后,机器记录下添加了哪一种碱基。Illumina公司从推出NextSeq和后来的MiniSeq,又推出了一种新的双色测序化学反应方法。与之前的区别在于核苷酸荧光修饰上,新的方法使用两种中一种颜色(红色或绿色)、无颜色(“黑色”)或同时结合两种颜色(红色和绿色的混合后呈现橙色) 来判定碱基种类。
一旦脱氧核糖核酸链被读取完成,刚刚合成的链就会被洗掉。然后,索引1引物进行结合,合成并测序索引1序列,然后被洗掉。该链再次形成一个桥,并且其3’端连接到流动池上的寡核苷酸上。索引2引物结合,合成并测得索引2的序列,然后被洗掉。
聚合酶在拱形链上合成互补链。它们由于变性而分开,每条链的3’端被封闭。正向链被洗掉,反向链重复边合成边测序的过程。
2.6 数据分析
测序同时发生在数百万个簇上,每个簇有大约1000个相同的脱氧核糖核酸插入片段的拷贝。[9]序列数据的分析是通过寻找具有重叠区域(称为重叠群)的片段,并将它们排列起来。如果参考序列是已知的,将重叠群与参考序列进行比较,以识别突变位点。
尽管从未得到过完整无断的序列,但这种获取零碎短片段的方法让科学家能够得到完整的序列;然而,由于Illumina读取长度不是很长[10]( HiSeq测序可以产生大约90 bp长的读取结果),[6]解决短串联重复序列可能是一个困难。[6][9]此外,如果序列是从头测序,参考基因组不存在,重复序列在序列组装中会造成很多困难。[9]还有些其他困难包括不精确的聚合酶反应、嵌合序列和PCR偏好性引起的碱基替换(特别是在读取结果的3’末端),[10]所有这些都可能导致产生不正确的序列。[10]
参考文献
- [1]
^Canard, Bruno; Sarfati, Simon (13 Oct 1994), Novel derivatives usable for the sequencing of nucleic acids, retrieved 2016-03-09.
- [2]
^Canard, Bruno; Sarfati, Robert S. (1994-10-11). "DNA polymerase fluorescent substrates with reversible 3′-tags". Gene. 148 (1): 1–6. doi:10.1016/0378-1119(94)90226-7. PMID 7523248..
- [3]
^"History of Illumina Sequencing". Archived from the original on 12 October 2014..
- [4]
^"Illumina - Sequencing and array-based solutions for genetic research". www.illumina.com..
- [5]
^Meyer, M.; Kircher, M. (2010). "Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing". Cold Spring Harbor Protocols. 2010 (6): pdb.prot5448. doi:10.1101/pdb.prot5448. PMID 20516186..
- [6]
^Feng, Y.-J., Q.-F., Chen, M.-Y., Liang, D. and Zhang, P. (2015). "Parallel tagged amplicon sequencing of relatively long PCR products using the Illumina HiSeq platform and transcriptome assembly". Molecular Ecology Resources. 16 (1): 91–102. doi:10.1111/1755-0998.12429. PMID 25959587..
- [7]
^Illumina, Inc. "Multiplexed Sequencing with the Illumina Genome Analyzer System" (PDF). Retrieved 25 September 2015..
- [8]
^Quail, Michael A (2012). "A Tale of Three next Generation Sequencing Platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq Sequencers". BMC Genomics. 13: 341. doi:10.1186/1471-2164-13-341. PMC 3431227. PMID 22827831..
- [9]
^Morozova, Marra O (Nov 2008). "Applications of next-generation sequencing in functional genomics". Genomics. 92 (5): 255–64. doi:10.1016/j.ygeno.2008.07.001. PMID 18703132..
- [10]
^Jeon, YS, Park SC, Lim J, Chun J, Kim BS (January 4, 2015). "Improved pipeline for reducing erroneous identification by 16S rRNA sequences using the Illumina MiSeq platform". Journal of Microbiology. 53 (1): 60–9. doi:10.1007/s12275-015-4601-y. PMID 25557481..
- [11]
^Pettersson E .,Lundeberge J .,Ahmadian A. (2008年)。测序技术的产生。基因组学”,第105-111页。.
- [12]
^Wang, Z; Fang, B; Chen, J; Zhang, X; Luo, Z; Huang, L; Chen, X; Li, Y (Dec 24, 2010). "De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas)". BMC Genomics. 11: 726. doi:10.1186/1471-2164-11-726. PMC 3016421. PMID 21182800..
- [13]
^Hao, Da Cheng; Ge, GuangBo; Xiao, PeiGen; Zhang, YanYan; Yang, Ling; Ellegren, Hans (22 June 2011). "The First Insight into the Tissue Specific Taxus Transcriptome via Illumina Second Generation Sequencing". PLoS ONE. 6 (6): e21220. doi:10.1371/journal.pone.0021220. PMC 3120849. PMID 21731678..
暂无